Chapter: HHT · Osler’s disease
Article: 2 of 13
Update: Mai 06, 2021
Author(s): Tinschert, Sigrid | Kühnel, Thomas
Hereditary hemorrhagic teleangiectasia is an autosomal-dominant hereditary disease. According to current knowledge, mutations in three different genes are causative. Mutations in the ENG (endoglin) gene (on chromosome 9) result in HHT type 1 (approx. 25-60%), mutations in the ACVRL1 gene (on chromosome 12) result in HHT type 2 (40-60%). Very rarely, mutations in the GDF2/BMP9 gene (on chromosome 10) result in HHT type 5. The existence of two other types, HHT type 3 and HHT type 4, is based only on genetic linkage studies; disease-causing genes have not yet been identified for them. A special form is the combination of hemorrhagic teleangiectasia (HT) with juvenile polyposis (JP). This disease (JPHT) is based on mutations of the tumor suppressor gene SMAD4 (on chromosome 18) and affects approximately 1% to 2% of patients with HHT.
All involved genes encode proteins that are important in the TGF-beta (ß) signaling pathway. A number of cellular functions of angiogenesis emanate from this signaling pathway. The proteins are expressed in endothelial cells.
Testing of the ENG, ACVRL1, and SMAD4 genes can detect the genetic cause in approximately 75% of individuals with HHT.
If the mutation in an HHT family is known, it is possible to search specifically for carriers, even among family members who are still asymptomatic. This opens up the opportunity of diagnosing clinically inapparent but complication-prone pAVM (pulmonary arteriovenous malformations) and cAVM (cerebral arteriovenous malformations). Such predictive diagnostics may (according to the Gene Diagnostics Act) only be initiated in the context of a genetic consultation by a specialist in human genetics.
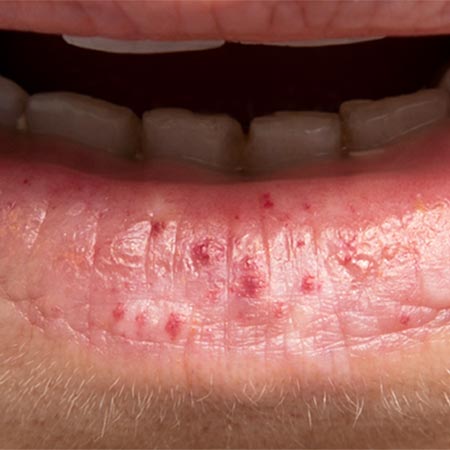
There is a certain genotype-phenotype relationship: there is a higher prevalence of pulmonary AVM (pAVM) and cerebral vascular malformations (CVM) in HHT type 1 (ENG) than in HHT type 2. In turn, vascular malformations of the liver and pulmonary hypertension are more frequently present in HHT type 2 (ACVRL mutations). In the special form JPHT (SMAD4), polyposis of the intestine is usually clinically in the foreground. The most common HHT features are epistaxis (cutaneous teleangiectasias may be absent) and pAVM.
Development of the vascular lesions at specific sites of the nasal mucosa cannot be explained by the genetic defect (germline mutation) alone, which is present in all body cells. The formation of the vascular lesions that ultimately bleed requires a further step in addition to the germline mutation; this is known as the “second hit” in the endothelial cells. Recently, it was shown that the second hit can be a mutation event affecting the second allele of the corresponding gene. The consequence is biallelic complete loss of function of the protein product in the affected endothelial cells. Whether other angiogenic triggers can also act as second hits, as has been suspected for a long time, has not yet been conclusively clarified.