Chapter: Vascular malformations associated with other anomalies
Article: 11 of 13
Update: Feb 02, 2021
Author(s): Tinschert, Sigrid | Wohlgemuth, Walter A.
Parkes Weber syndrome (PWS) is characterized by the combination of cutaneous capillary malformations (CM) and arteriovenous malformations (AVM) and multiple very fine arteriovenous fistulas (AVF) with an ipsilateral circumscribed overgrowth of an extremity. Lymphatic malformations (LM) may also be present and appear as lymphedema.
Parkes Weber syndrome is usually sporadic (non-familial), with rare familial occurrence (paradominance). It is caused by loss-of-function mutations in the RASA1 gene, which encodes the p120-Ras-GTPase activating protein (p120-RasGAP) and converts active Ras, bound to GTP, to the inactive, GDP-bound form. Vascular malformation presumably results only from complete loss of function of RASA1 due to biallelic mutations (LOH = “loss of heterozygosity”). The segmental changes (localized vascular malformations and overgrowth) would be explainable by an early embryonic LOH of the RASA1 gene against the background of an inherited constitutional (present in all cells) RASA1 mutation and correspond to a so-called type 2 mosaic (according to Happle: syndrome of type 2 segmental nevus rhodoides). Multiple CM/AVM without overgrowth symptoms represent an oligosymptomatic variant and can be explained by later (non-early embryonic) RASA1 mutations.
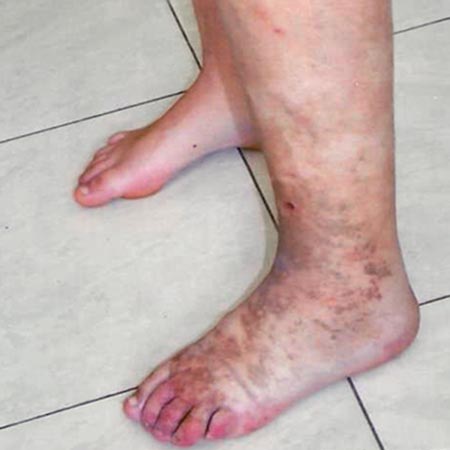
Regional overgrowth (subcutaneous adipose tissue, muscle, and bone) usually affects one limb or parts of it. The legs are more commonly affected than the arms.
Capillary malformations (CM) and small arteriovenous shunts (micro-AVM/AVF) located in the subcutis result in the typical appearance of pink to red (rhodoid) nevi of PWS. The nevi are of variable size and number, with at least one large nevus always present. Smaller nevi are clustered with a halo surrounding them. The affected skin region is hyperthermic because of shunt-induced increased perfusion.
Microfistulous AVM/AVF can be found not only in the subcutis but also in the muscles and bones.
Lymphatic malformations may also occur and manifest as lymphedema.