Chapter: Venous malformations
Article: 12 of 13
Update: Mar 25, 2021
Author(s): Barbera, Letterio Christian
These include the segmental overgrowth syndromes as well as the Servelle-Martorell and Maffucci syndromes. In addition to venous malformations, the patients show associated circumscribed and asymmetric anomalies of connective tissue and skeletal system.
Proteus syndrome, CLOVES syndrome and Klippel-Trénaunay syndrome are characterized by a somatic mutation that is not inherited and is only detectable in the affected part of the body (genetic mosaic). As defined in the name, there is segmental overgrowth, usually affecting one side of the body (face, extremity). Differences in both circumference and length vary during growth, but their extent cannot be predicted. There are no detailed studies of the natural course of the growth, but the impression is that the overgrowth is more pronounced in the first growth phase (2–4yrs) than in the third (11–14yrs).
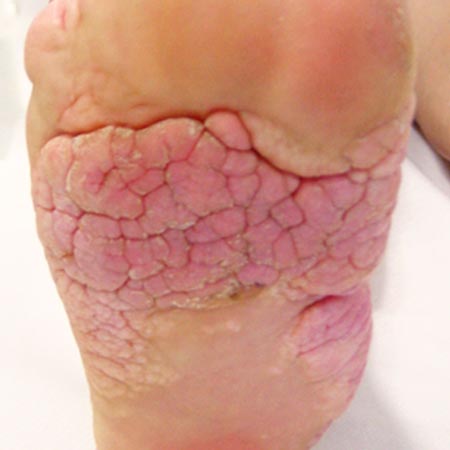
Asymmetric, often gigantic bone overgrowth is first noticed after birth, increases disproportionately during the growth phases and can affect the skull, trunk and extremities. The proportions of the bone can be so altered that the original shape is no longer recognizable. The subcutis can be thicker or thinner on the affected parts of the body compared to the contralateral side. Benign skin tumors with a surface similar to the surface of the brain are pathognomonic for this syndrome (so-called cerebriform mixed connective tissue nevus). The associated vascular malformations usually occur as combined slow-flow malformations (LVM, CVM, CLVM).
Circumscribed adipose tissue proliferation, congenital vascular malformations (VM, CM, LM and AVM) and epidermal nevi, which can occur on the head, trunk and extremities, characterize this overgrowth syndrome. Despite the benign nature of the disease, the conspicuous disfigurement of the affected body part causes relevant impairment of the quality of life, which can be partially corrected by plastic surgery. The risk of thromboembolic complications is high.
Klippel-Trénaunay syndrome (KTS) in the strict sense is the first congenital vascular malformation to have been described, although the term is often mistakenly used for other vascular malformations.
The following triad is characteristic of KTS: disproportional overgrowth of a limb or side of the body + capillary malformation (nevus flammeus) + venous malformation +/- lymphatic malformation. The overgrowth can be considerable and cause severe walking difficulties. It is therefore an anomaly that severely affects the life of an affected person.
Contrary to earlier assumptions, it is not the malformed vessels but concomitant primary dysplasia of the soft tissue that is responsible for the overgrowth. The vascular anomalies extend over the affected body side with a predilection for the lateral parts. While the capillary malformation may become less conspicuous and is sometimes only recognizable from the reticular veins that have developed secondarily as outflow vessels, the venous malformation increases in size over the years. Subcutaneous vessels of large caliber on the outside of the leg enter the deep venous system at the knee, thigh or gluteal level. Although this venous malformation is referred to as a marginal vein or embryonic vein when there is aplasia of the deep veins, the existing vascular morphology often corresponds more to a venous plexus than a singular vessel.
These findings are relevant for therapy planning and prognosis. The indication for an early, extensive resection of the venous malformation must therefore be critically examined. Consistent compression and exercise-based therapy with selective treatment of painful areas seems to be the better alternative. Communications with the deep venous system should be closed because of the risk of thromboembolism.
PTEN hamartoma syndrome comprises a heterogeneous group of rare diseases with segmental overgrowth, soft tissue tumors and slow-flow and sometimes fast-flow vascular malformations. The neoplasms can occur on the skin, mucous membranes of the colon, mammary gland and intestinal tract and are considered precancerous. The common feature of this syndrome group is the presence of an autosomal dominant germline mutation of the PTEN gene, which is detectable in the blood, and a so-called second hit (local mutation of the other PTEN-coding allele) in the affected tissue. The associated venous malformation plays a clinically less relevant role than the often accompanying AVM. It is important to remember that such a syndrome may be present when parts of a venous malformation or AVM are embedded in particularly large, usually fatty soft tissue masses.
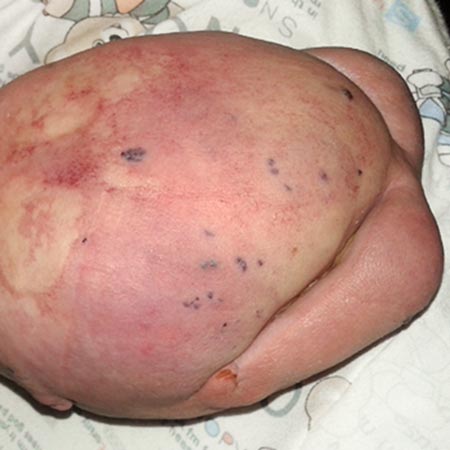
Bean syndrome is a rare sporadic disorder characterized by venous malformations of the skin, mucous membranes and internal organs. The multifocal small lesions are livid to dark blue in color, slightly raised and often only a few mm in size (“bleb”). The surface has a rubber-like consistency, fades under pressure and is rarely painful. The cutaneous manifestation occurring in childhood is clinically irrelevant except for the appearance. Later, these vascular changes proliferate de novo, occur in the internal organs and become conspicuous by recurrent bleeding. The gastrointestinal tract (GIT) is most frequently affected, where both acute and chronic blood loss occurs. This is manifests like any other GI bleeding (hematemesis, melena, anal blood loss) and can become life-threatening. Chronic blood loss leads to iron deficiency anemia and fatigue. Treatment consists of blood transfusions, iron replacement, endoscopic coagulation (e.g. by laser), often with endoscopically controlled sclerotherapy, and, if this is not possible, partial resection of the affected intestinal segment. Control endoscopies are the most important preventive measure after diagnosis. Other, albeit rarer, extracutaneous localizations are in bones, brain and abdomen, where cerebral hemorrhages, pathological fractures and severe menorrhagia can occur. The volume of the malformations increases during the last trimester so that there is an increased risk of bleeding during birth. In that case, appropriate multidisciplinary care of the pregnant woman is necessary.
In this special form, the venous malformation occurs together with partial hypoplasia of one limb. The etiology of the syndrome is unknown. There are noticeable differences in circumference, for which hypoplasia of both the subcutaneous fat and the muscle tissue are responsible. Small differences in the length of the tubular bones cause a length discrepancy and cannot be compensated for by orthopedic surgery. Interventions to slow down the growth of the contralateral leg are usually not indicated. Treatment options such as physical training and shoe height compensation are symptomatic in nature, but should nevertheless be implemented. In selected cases, interventions may be necessary.
This rare disease with bone and vascular anomalies, often affecting the upper extremity, is sporadic in most cases and may be associated with a heterozygous autosomal dominant germline mutation. The age of manifestation is 4–5 years and the course is usually mild. In addition to large-lumen, venous malformations, multiple enchondromas occur in different locations, resulting in axial deformities and conspicuous bulging of long bones. These enchondromas can go through malignant transformation in the long term and must be monitored. Pathological fractures may occur, so that appropriate precautions are recommended. Treatment is based on the symptoms; esthetic corrective procedures should only be done if strictly indicated because of increased complication rates (fracture, bleeding).